ناشنوایی عارضهای شایع در میان تمام جمعیت ها بوده به نحوی که گفته می شود سالانه از هر 1000 نوزاد متواد شده، دو تا سه نوزاد دارای مشکل در شنوایی میباشند. این عارضه که میتواند افراد را از بدو تولد تا سنین بالای 75 سال درگیر نماید، عمدتاً بر اساس متوسط میزان نشنیدن اصوات از خفیف تا بسیار شدید تقسیم بندی می گردد.
انواع ناشنوایی:
- ناشنوایی خفیف; متوسط میزان نشنیدن اصوات در این حالت از 16 تا 25 دسیبل است.
- ناشنوایی ملایم; متوسط میزان نشنیدن اصوات در این حالت از 26 تا 40 دسیبل است.
- ناشنوایی متوسط; متوسط میزان نشنیدن اصوات در این حالت از 41 تا 55 دسیبل است.
- ناشنوایی کمی شدید; متوسط میزان نشنیدن اصوات در این حالت از 56 تا 70 دسیبل است.
- ناشنوایی شدید; متوسط میزان نشنیدن اصوات در این حالت از 71 تا 90 دسیبل است.
- ناشنوایی بسیار شدید; متوسط میزان نشنیدن اصوات در این حالت از 91 دسیبل به بالا است.
مطالب دیگر را بخوانید: تغذیه در بیماران فنیل کتونوری
ناشنوایی
عوامل دخیل در بروز ناشنوایی:
- علل ژنتیکی; بیش از 60 درصد از ناشنواییها به علت نقایص ژنتیکی ایجاد میگردند.
- علل غیر ژنتیکی;
- افزایش سن. در انسان به تدریج با افزایش سن توانایی شنیدن صداها با فرکانس بالا کاهش مییابد.
- قرار گیری در معرض آلودگیهای صوتی
- عفونتهای مادرزادی، به ویژه آلودگی مادرزادی بهCytomegalovirus (CMV).
- ابتلا به بیماریهایی نظیر MS، مننژیت، سفلیس و عفونتهای باکتریایی.
- استفاده از داروهای اتوتوکسیک نظیر آمینوگلیکوزیدها و سیکلوفسفامید.
- مصرف الکل در دوران بارداری. مصرف الکل توسط مادران باردار میتواند سبب آسیب شنوایی در جنین گردد.
- آسیبهای فیزیکی
ناشنواییهای دارای علل ژنتیکی به دو دسته ناشنواییهای سندرمیک و ناشنواییهای غیر سندرمیک تقسیم بندی میگردند.
ناشنواییهای سندرمیک:
این دسته از ناشنواییها حدود 30 درصد از ناشنواییهای دارای علل ژنتیکی را تشکیل میدهند و علاوه بر فنوتیپ ناشنوایی دارای علائم و نشانههای دیگری در سایر ارگانهای بدن میباشند. از این جمله میتوان به سندرم Pendred، سندرم Usher، سندرم Waardenburg و سندرم Branchiootorenal اشاره نمود.
ناشنواییهای غیر سندرمیک:
این دسته از ناشنواییها حدود 70 درصد از ناشنواییهای دارای علل ژنتیکی را تشکیل میدهند و خود به سه زیر گروه تقسیم بندی میگردند:
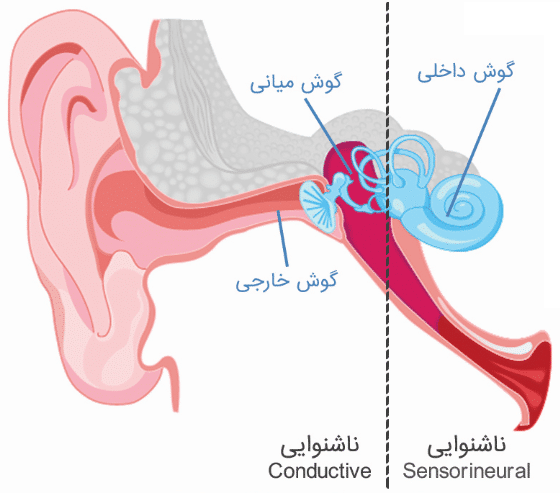
- ناشنوایی Sensorineural؛ با آسیب به گوش داخلی
- ناشنوایی Conductive؛ با آسیب به گوش میانی
- ناشنوایی Mixed؛ با آسیب به گوش میانی و داخلی
مطالب دیگر را بخوانید: دیستروفی میوتونی
ناشنوایی
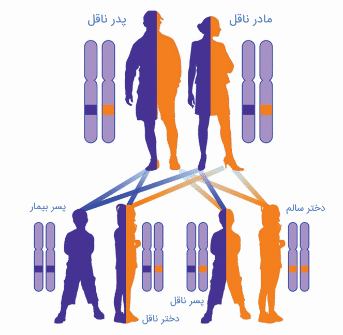
- ناشنواییهای دارای علل ژنتیکی طیفی از توارثهای اتوزومال غالب و مغلوب، توارث وابسته به ایکس و توارث میتوکندریایی را بروز میدهند.
- در ناشنواییهای غیر سندرمیک، بیش از 80 درصد از موارد دارای توارث اتوزومال مغلوب میباشند. 15 درصد الگوی توارثی اتوزومال غالب را بروز میدهند و دو الگوی توارثی وابسته به ایکس و میتوکندریایی هر کدام دارای سهمی در حدود یک درصد میباشند.
ناشنوایی یک بیماری به شدت هتروژن میباشد، به گونهای که در حالت غیرسندرمیک این بیماری با بیش از 100 ژن دارای همبستگی بوده و درحالت سندرمیک نیز با بیش از 400 سندرم ژنتیکی مرتبط میباشد که هر یک از این سندرمها ممکن است با یک یا چندین ژن پیوستگی داشته باشند.
ناشنوایی های غیر سندرمیک
- ناشنواییهای غیرسندرمیک با الگوی توارث اتوزومال مغلوب در50 درصد از موارد در یکی از دو ژن GJB2 و GJB6 که در لوکوس DFNB1 واقع شدهاند دارای جهش میباشند. این جهشها عمدتاً در ژن GJB2 واقع شدهاند، به گونهای که تاکنون بیش از 150 تغییر بیماریزا در این ژن شناسایی شده است. در میان این تغییرات، سه جهش 35delG، 167delT و235delC شایعترین موتاسیونهای شناخته شده در ژن GJB2 میباشند. همچنین در ناشنواییهای غیرسندرمیک که موتاسیون در لوکوس DFNB1 واقع شده است، ممکن است یک جهش در ژن GJB2 در یک آلل و یک حذف در ژن GJB6 در آلل دیگر باشد و یا آنکه حذف در دو آلل مربوط به ژن GJB6 ایجاد گردد.
- ناشنواییهای غیرسندرمیک با الگوی توارث اتوزومال غالب میتوانند به واسطه جهشهای غالب در ژن GJB2 ایجاد شوند ولی این موضوع متداول نیست.
- ناشنواییهای غیرسندرمیک با الگوی توارث میتوکندریایی که در طیف متوسط تا شدید دسته بندی میشوند، با موتاسیونها در دو ژن MT-RNR1 و MT-TS1 همبستگی دارند.
چندین ژن و لوکوس مستعد کننده دیگر نیز مانند KCNQ4، GRHL2، MYO15A، CDH23،PCDH15 و… برای ناشنوایی غیرسندرمیک شناسایی شده است ولی با این حال در درصد قابل ملاحظهای از بیماران هنوز علت ژنتیکی بیماری نامشخص میباشد.
ناشنوایی های سندرمیک
برای هریک از سندرمهایی که در آنها ناشنوایی مشاهده میشود یک یا چندین ژن شناسایی شده است. از این جمله میتوان به موارد زیر اشاره نمود:
- شناسایی ژن SLC26A4 برای سندرم Pendred
- شناسایی ژن PAX3 برای سندرم Waardenburg نوع یک
- شناسایی ژنهای MITF و SOX10 برای سندرم Waardenburg نوع دو
- شناسایی ژنهای MYO7A و USH2A به ترتیب برای سندرم Usher نوع یک و نوع دو
- شناسایی ژن EYA1 برای سندرم Branchiootorenal نوع یک
توارث بیماری
- ناشنواییهای ناشی از علل ژنتیکی میتوانند به صورت توارثهای اتوزومال غالب و مغلوب، توارث وابسته به ایکس و توارث میتوکندریایی بروز کنند.
- در ناشنواییهای غیر سندرمیک، بیش از 80 درصد از موارد دارای توارث اتوزومال مغلوب میباشند. ۱۵ درصد از ناشنواییهای ژنتیکی به صورت الگوی توارثی اتوزومال غالب بروز میدهند، در حالی که هر کدام از الگوهای توارث وابسته به ایکس و میتوکندریایی سهمی در حدود یک درصد دارند.

ژنتیک بیماری
ناشنوایی یک بیماری به شدت هتروژن میباشد، به گونهای که در حالت غیرسندرمیک این بیماری با بیش از 100 ژن دارای همبستگی بوده و درحالت سندرمیک نیز با بیش از 400 سندرم ژنتیکی مرتبط میباشد که هر یک از این سندرمها ممکن است با یک یا چندین ژن پیوستگی داشته باشند.
ناشنواییهای غیر سندرمیک
- ناشنواییهای غیرسندرمیک با الگوی توارث اتوزومال مغلوب در50 درصد از موارد در یکی از دو ژن GJB2 و GJB6 که در لوکوس DFNB1 واقع شدهاند دارای جهش میباشند. این جهشها عمدتاً در ژن GJB2 رخ میدهند و تاکنون بیش از 150 تغییر بیماریزا در این ژن شناسایی شده است. از میان این تغییرات، سه جهش 35delG، 167delT و 235delC به عنوان شایعترین موتاسیونهای شناختهشده در ژن GJB2 شناخته میشوند. همچنین، در ناشنواییهای غیرسندرمیک که موتاسیون در لوکوس DFNB1 قرار دارد، ممکن است یک جهش در ژن GJB2 در یک آلل و یک حذف در ژن GJB6 در آلل دیگر مشاهده شود، یا اینکه حذف در هر دو آلل مربوط به ژن GJB6 اتفاق بیفتد.
- ناشنواییهای غیرسندرمیک با الگوی توارث اتوزومال غالب میتوانند به واسطه جهشهای غالب در ژن GJB2 ایجاد شوند ولی این موضوع متداول نیست.
- ناشنواییهای غیرسندرمیک با الگوی توارث میتوکندریایی که در طیف متوسط تا شدید دسته بندی میشوند، با موتاسیونها در دو ژن MT-RNR1 و MT-TS1 همبستگی دارند.
چندین ژن و لوکوس مستعد کننده دیگر نیز مانند KCNQ4، GRHL2، MYO15A، CDH23،PCDH15 و… برای ناشنوایی غیرسندرمیک شناسایی شده است ولی با این حال در درصد قابل ملاحظهای از بیماران هنوز علت ژنتیکی بیماری نامشخص میباشد.

تشخیص و مشاوره ژنتیک
تشخیص انواع ناشنوایی و دلایل ژنتیکی بروز آن از جهات زیر دارای اهمیت میباشد:
- تعیین نوع آسیب سیستم شنوایی
- تعیین ثابت ویا پیشرونده بودن بیماری
- انتخاب روشهای درمانی مناسب، مانند استفاده از سمعک یا کاشت حلزون شنوایی، بر اساس نوع تغییر ژنتیکی و آسیب وارد شده اهمیت زیادی دارد. به عنوان مثال، افرادی که در ژن GJB2 دارای جهش هستند، میتوانند از این روشهای درمانی بهرهمند شوند. این افراد دارای ناشنوایی از نوع Sensorineural بوده و معمولاً فنوتیپ ناشنوایی را در بدو تولد به صورت دو طرفه و غیر پیشرونده بروز میدهند. کاشت حلزون شنوایی برای این دسته از بیماران یکی از بهترین روشهای درمانی بوده و این افراد معمولاً بهترین پاسخ را به درمان با کاشت حلزون شنوایی نشان میدهند.
- استفاده از خدمات مشاوره ژنتیک به همراه به کارگیری تستهای تشخیصی در مراحل پیش از بارداری و همچنین حین بارداری در خانوادههایی که دارای فرزند ناشنوا بوده و یا یک یا هر دو والد ناشنوا هستند، میتواند از انتقال بیماری جلوگیری نموده و موجب تولد فرزندانی سالم و شنوا گردد.

دستورالعمل American College of Medical Genetics (ACMG) در مواجه با بیماران مبتلا به ناشنوایی
- مراجعه افراد با مشکلات شنوایی به متخصصین مربوطه. چناچه بررسیهای اولیه نشان دهد که علت بیماری فرد میتواند به دلیل وجود نقایص ژنتیکی باشد، بایستی فرد به مشاور ژنتیک ارجاع یابد.
- مراجعه به مشاور ژنتیک. مشاور ژنتیک با بررسی تستهای شنوایی، علائم فرد و سابقه بیماری در او و خانوادهاش، به تعیین سندرمیک یا غیر سندرمیک بودن ناشنوایی بیمار میپردازد.
- تشخیص سندرمیک بودن ناشنوایی. در این صورت باید ژن(های) سندرم مورد نظر در فرد بیمار مورد بررسی قرار گیرد.
- تشخیص غیر سندرمیک بودن ناشنوایی. در این صورت ابتدا باید دو ژن GJB2 و GJB6 در فرد بیمار مورد بررسی قرار گیرد.
بررسی ژن GJB2 به واسطه انجام واکنش PCR و به دنبال آن توالی یابی این ژن انجام میگیرد. بررسی ژن GJB6 نیز به دلیل وجود
حذفهای شایع در این ژن به کمک تکنیک GAP-PCR صورت میگیرد.
از آنجایی که جهشهای ژن GJB2 تنها عامل حدود ۱۸ درصد از موارد ناشنوایی در جمعیت ایرانی هستند و جهش در ژن GJB6 نیز به ندرت در این جمعیت مشاهده میشود، در صورت عدم شناسایی جهش در این دو ژن، باید با استفاده از روش Next-Generation Sequencing (NGS) به بررسی سایر ژنهای دخیل در ناشنوایی پرداخت. برای تعیین علت ژنتیکی ناشنوایی، میتوان از دو روش استفاده کرد: Targeted Exome Sequencing که از پنلهایی استفاده میکند که تنها ژنهای شناختهشده مرتبط با ناشنوایی را بررسی میکنند، و Whole Exome Sequencing که تمامی ژنهای فرد بیمار را مورد بررسی قرار میدهد. در آزمایشگاه ویرا، شناسایی ناشنوایی ژنتیکی با دقت و صحت بسیار بالا انجام میشود.
تشخیص انواع ناشنوایی و دلایل ژنتیکی بروز آن از جهات زیر دارای اهمیت میباشد:
- تعیین نوع آسیب وارد شده به سیستم شنوایی
- تعیین ثابت ویا پیشرونده بودن بیماری
- اتخاذ روشهای درمانی مناسب اعم از استفاده از سمعک، کاشت حلزون شنوایی و … بر اساس نوع تغییر ژنتیکی و آسیب وارد شده. بهترین نمونه در این زمینه افرادی هستند که در ژن GJB2 دارای جهش میباشند. این افراد دارای ناشنوایی از نوع Sensorineural بوده و معمولاً فنوتیپ ناشنوایی را در بدو تولد به صورت دو طرفه و غیر پیشرونده بروز میدهند. کاشت حلزون شنوایی برای این دسته از بیماران یکی از بهترین روشهای درمانی بوده و این افراد معمولاً بهترین پاسخ را به درمان با کاشت حلزون شنوایی نشان میدهند.
- استفاده از خدمات مشاوره ژنتیک به همراه به کارگیری تستهای تشخیصی در مراحل پیش از بارداری و همچنین حین بارداری در خانوادههایی که دارای فرزند ناشنوا بوده و یا یک یا هر دو والد ناشنوا هستند، میتواند از انتقال بیماری جلوگیری نموده و موجب تولد فرزندانی سالم و شنوا گردد.
آزمایشگاه ژنتیک ویرا با بهرهگیری از روشهای پیشرفته مانند توالییابی نسل جدید (NGS) و آزمایشهای هدفمند ژنتیکی، به تشخیص دقیق علت ژنتیکی ناشنوایی میپردازد. با بررسی ژنهای مرتبط همچون GJB2 و GJB6، امکان شناسایی جهشهای مؤثر فراهم شده و برنامههای مشاوره و درمان متناسب مانند کاشت حلزون شنوایی پیشنهاد میگردد. خدمات مشاوره ژنتیک ویرا میتواند به خانوادهها در پیشگیری از انتقال ناشنوایی به نسل بعد کمک کند.
مطالب دیگر را بخوانید: ناشنوایی ژنتیکی به دلیل جهش GJB2; تشخیص و پیشگیری